一、前言
新南向國家中以印度人口最多,為全球第二大人口國家,僅次於中國大陸,2016年總人口數為13.2億。印度人均GDP約1,500美元,在新南向國家中屬後段,但因龐大人口數,印度醫療器材市場反而是最大的。特別是印度國內醫材市場高度仰賴進口,現階段進口醫療器材約占印度整體醫療器材市場7成以上,並以美、日廠商為主,目前我國出口至印度之醫療器材品項,則以血糖計、導管耗材、醫用塑膠產品等品項為主力產品。
因應新法規之修正,印度自2018年起針對醫療器材採取風險分級管理新制,將醫療器材與藥品管理機制加以分離、簡化,同時,在上市查驗登記程序中導入第三方驗證機構,針對就醫療器材之規格及品質進行驗證(certify),有助於吸引相關領域投資及整體醫療器材市場的健全發展。依據印度醫療器材產業成長趨勢之預估,顯示印度各類別醫材產值至2020年均有13%以上之成長幅度,其中,耗材與植入體之成長幅度最高,預估2020年(相較於2014年)之成長幅度達到19%。
醫療器材為我國生技醫療產業之主力,研發量能較高的牙材、骨材、智慧醫療等產品已經逐漸成為我國出口潛力產品,相較其他先進國家在成本效益方面更具優勢,顯示我國醫療器材在新南向市場具有較佳之發展潛力。本文從印度2018年醫材法規修法背景與修正重點出發,整體性介紹新制實施後現階段印度醫材監管法規之制度全貌。
二、2018年醫材法規修正重點
2015年印度政府公布「國家醫療器材政策綱領」,計畫修訂醫療器材管理法規,改革醫療器材上市查驗登記程序與管理方式。基於此一政策方向,印度政府在2017年1月公布新版《醫療器材管理規則》(Medical Device Rules 2017),本次修法之重點有二:一是將醫療器材與藥品管理機制加以分離、簡化;二是在查驗登記程序中導入第三方驗證機構,針對就醫療器材之規格及品質進行驗證(certify),有助於吸引相關領域投資及整體醫療器材市場的健全發展。
除此之外,新版《醫療器材管理規則》也針對上市查驗登記制度做出重大修正,包括:簡化醫療器材業者取得許可證之查驗程序、放寬實施臨床試驗之要求、針對政府部門依法採行之行政措施設置明確的時限規定,並導入可透過電子傳輸方式提交申請資料之數位化申請平台。目前新版《醫療器材管理規則》已於2018年1月1日起生效實施。
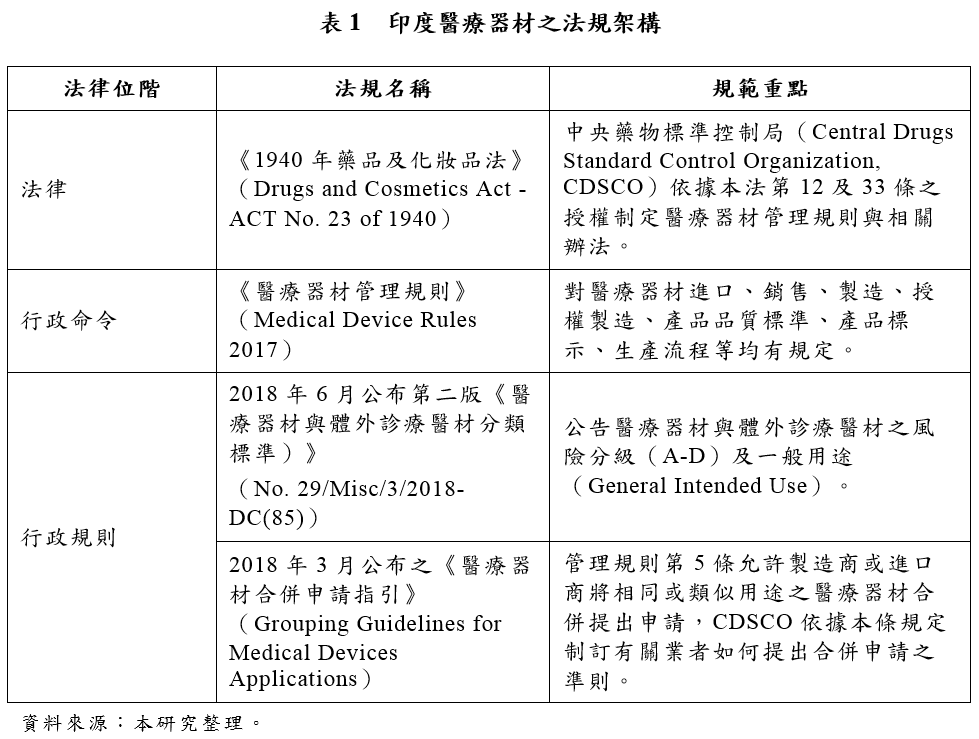
三、醫材監管相關主管機關(一)中央藥物標準控制局(Central Drugs Standards Control Organization, CDSCO) 印度健康及家庭福利部轄下之「中央藥物標準控制局」(Central Drugs Standard Control Organization, CDSCO),為印度醫療器材之中央主管機關,其內部以「印度藥物管控聯合委員會」(Drugs Controller General of India, DCGI)作為同時監管全國藥政與醫療器材之最高決策單位,主導藥品、醫療器材等產品相關政策制定,並依法審核相關業者之申請。
2018年新版醫材管理規則實施後,分類為C級(中高風險)、D級(高風險)風險等級較高之醫療器材,其製造與進口皆須取得中央醫藥監管機關所核發之許可證。據此,CDSCO依法擔任中央醫藥監管機關之角色,主要負責C級(中高風險)、D級(高風險)醫療器材許可之審查與核發事項。除此之外,進口(所有類別)外國醫療器材仍由CDSCO負責審核與授予許可證。
(二)省級藥物管制局(State Drugs Controller) 2018年新版醫材管理規則實施後,各地方政府(省級)所設置之藥物管制局(State Drugs Controller)擔任低風險醫材之監理機關,以及上市許可之授證單位。其中,分類為A級(最低風險)之醫療器材依法採取自我管理,其流通、販售、使用無須事先向省級藥物管制局申請許可。然而,分類為A級(最低風險)、B級(較低風險)之低風險醫療器材,其製造、流通、販售、使用均屬省級藥物管制局之主要管轄範圍。
(三)國家認證委員會(National Accreditation Board for Certification Bodies, NABCB) 2018年新版管理規則在查驗登記程序中導入了第三方驗證機構評鑑制度,授權這些驗證機構針對醫療器材製造商的品質管理體系進行驗證和評估,由驗證機構執行醫材製造場所及產品稽查,以確保醫療器材品質符合印度法規標準之要求。為了確保驗證機構具備一定水準之驗證與評估能力,新法由印度國家認證委員會(National Accreditation Board for Certification Bodies, NABCB)擔任此一驗證體系之認證機構。經NABCB認可之驗證機構即可擔任新法所稱之「查核機構」(Notified body),始具備醫材品質管理體系之驗證與評估能力。
四、醫療器材監管範圍與分類方式(一)醫療器材定義
印度《1940年藥品及化妝品法》將本法所規範之醫療器材定義為:「包括在人類或動物身體內部或外部用於診斷,治療,緩解或預防病症的器材
[1]」。2017年公布之《醫療器材管理規則》第3條特別針對納入管理範圍之醫療器材做出更為明確之定義:「本規則所定義之醫療器材,係指所有使用於人類及動物體內或體外之特定器材、其目的在於診斷、治療、舒緩、預防疾病與障礙以及影響特定生理機能(如節育)」。相較於舊法公告列管之23項管制類醫療器材,新版管理規則擴大了醫療器材之監管範圍,進一步將下列醫療器材之列入管制範圍
[2]:
1. 經政府公告管制能夠影響人體功能或結構之醫療器材;
2. 外科敷料、手術繃帶、外科手術縫合線、手術縫合線、結紮線、血液和含有血液之收集袋;
3. 用於體外診斷(in vitro diagnosis)之醫療器材;
4. 所有具有診斷、治療、舒緩、預防人體或動物疾病與障礙等功能之醫療器材。
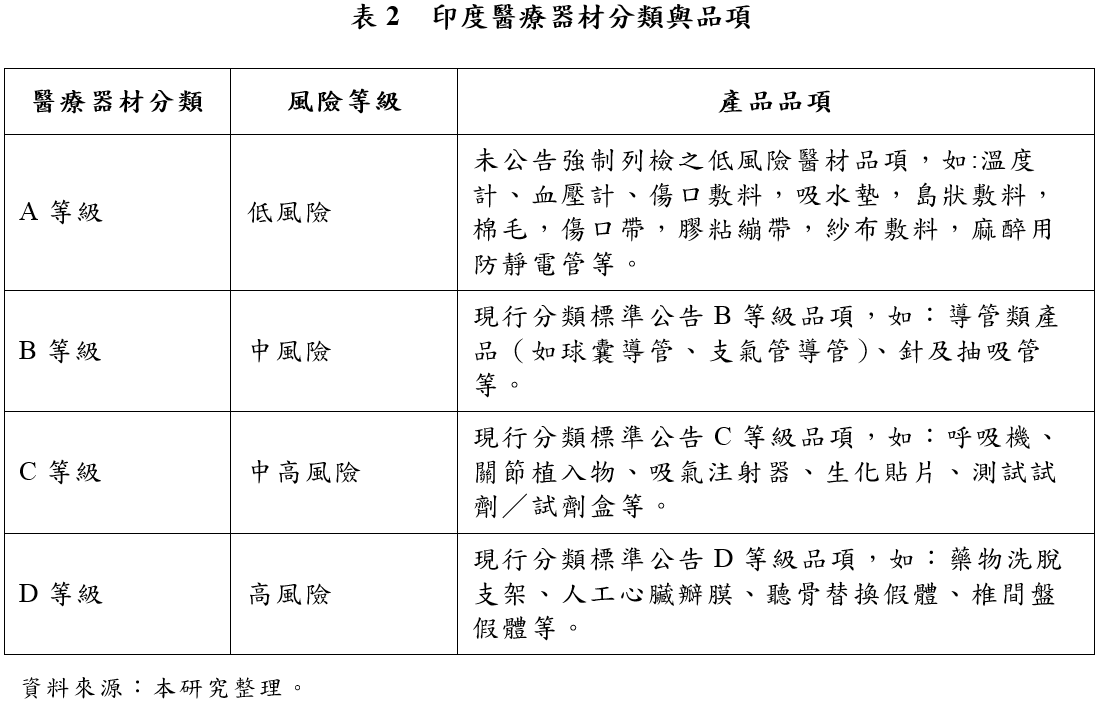
(二)醫療器材之分類 印度實施新版《醫療器材管理規則》之前(即2018年1月之前),醫療器材未明訂分類分級,僅公告需於上市前進行註冊之管制類醫療器材共23類。新版《醫療器材管理規則》係參考「全球醫療器材法規調和會」(Gobal Harmonization Task Force, GHTF)指引,採取風險分級管理原則將被管制之醫療器材劃分為A、B、C、D 四種等級。其中,A類醫療器材為低風險產品,風險程度依序增加至最高風險之D類產品。此外,體外診斷醫療器材亦將另行劃分為A~D四類。
印度CDSCO已經在2017年11月公布了《醫療器材與體外診療醫材分類標準》(No.29/Misc.l3/2017-DC(292)),公告現階段所有列管之醫療器材與體外診療醫材項目,包括各項產品之風險分級(A~D)以及一般用途(General Intended Use)。然而,在相較於其他國家給予業者提出登記申請時,於判斷產品分類方面具備一定程度之彈性,印度並未給予醫材進口商、製造商在判斷產品分類上之彈性空間,分類標準要求醫材業者在申請產品登記時,應遵循「印度藥物管控聯合委員會」(Drugs Controller General of. India, DCGI)針對個別醫療器材申請案之分類決定
[3]。對於未判斷分類之申請案件,依法均應先向CDSCO提出分類申請,如不服分類決定可以在30日內提出申請複查。
[4]
五、製造商申請上市查驗程序與要求
(一)透過單一電子化窗口提交申請文件
印度2018年新實施之醫材管理規則,針對不同風險等級之醫療器材制訂了不同程度之上市前產品查驗程序與審核文件要求,同時,對於進口醫療器材要求進口商應向CDSCO申請取得進口許可。然而,基於申請程序之電子化,舉凡進口、製造、銷售或配銷許可證之申請,以及產品臨床試驗之審查等,無論產品之審核權責機關屬於中央CDSCO抑或是省級藥物管制局,業者均可透過CDSCO所設置之單一電子化窗口(https://cdscomdonline.gov.in)提交申請書與相關文件。
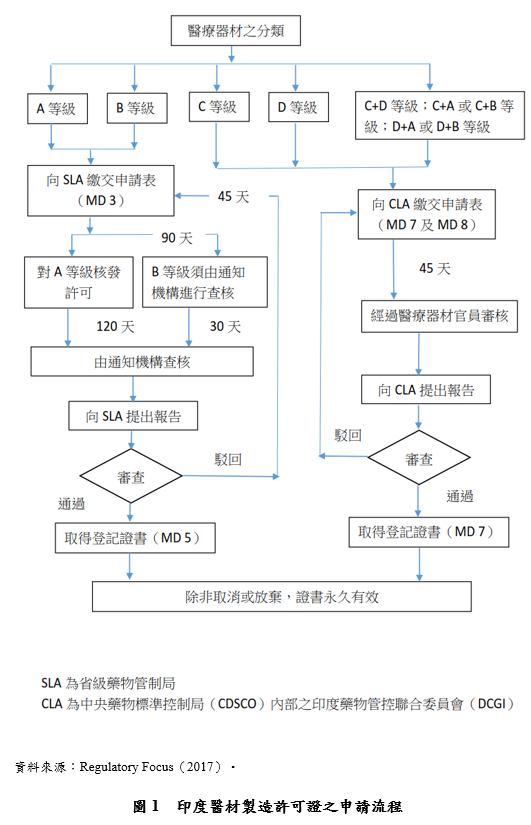
(二)申請A等級(低風險)、B等級(中風險)醫療器材之製造許可證
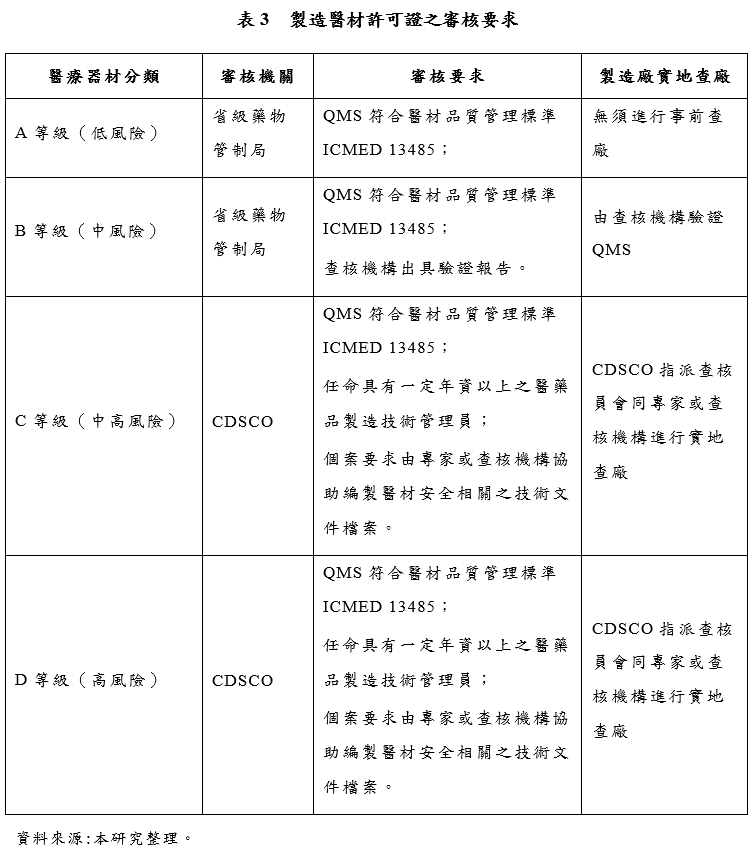
製造A等級(低風險)、B等級(中風險)醫療器材以供販售、流通使用或租賃之醫材製造商,依法應向省級藥物管制局申請製造許可證
[5]。其中,醫材製造商應提出製造廠之品質管理系統(Quality Management System, QMS)符合印度醫療器材品質管理標準ICMED 13485要求之證明文件
[6]。在申請A等級醫材製造許可證時,依法不需要在核發許可證之前進行實地查廠之程序
[7]。省級藥物管制局於審核程序中僅就製造商所提出之自我驗證文件進行審核,然而,在核發許可證後120日內,製造商依法必須由NABCB認可之查核機構進行實地查廠。確認製造廠之QMS符合印度醫療器材品質管理標準ICMED 13485之要求。
相對於此,申請B等級醫材製造許可證時,則需要進行事前實地查廠之程序。此時,管理規則要求申請許可證之製造商應實施第三方實地查廠程序,由NABCB認可之查核機構針對製造廠進行驗證,確認製造廠之QMS符合印度醫療器材品質管理標準ICMED 13485之要求。查核機構應在製造商提出許可證申請之90日內完成QMS驗證程序,並將驗證報告提交給省級藥物管制局進行審核。依法省級藥物管制局應在收受驗證報告書起20日內作出是否核發許可證之決定。
[8](三)申請C等級(中高風險)、D等級(高風險)醫療器材之製造許可證 製造C等級(中高風險)、D等級(高風險)醫療器材以供販售、流通使用或租賃之醫材製造商,依法應向CDSCO申請製造許可證
[9]。管理規則對於申請C等級、D等級醫療器材製造許可證之審核要求大致相同:
1. 要求製造廠之品質管理系統,必須符合印度醫療器材品質管理標準ICMED 13485;
[10] 2. 任命具有一定年資以上之醫藥品製造技術管理員;
[11] 3. 個案要求由專家或查核機構協助編製醫材安全相關之技術文件檔案。
[12] 相較於低風險醫材許可證授權第三方查核機構實施查廠之規定,管理規則要求申請C等級、D等級醫療器材製造許可證時,應由CDSCO指派醫療器材查核官員(Medical Device Officers)組成查核小組實施「到廠查核」(Inspection)。查核小組應由至少2名查核官員組成,得會同專家或NABCB認可之查核機構參與查核小組
[13]。查核小組應在CDSCO受理許可證申請起45日內實施到廠查核,完成查核報告後應提交給CDSCO進行審核。CDSCO在收受查核報告後45日內應做出是否核發許可證之決定。
[14]
六、進口許可證申請流程 境外醫療器材製造商如欲使其製造之醫療器材在印度可以流通使用或銷售,必須由授權印度當地代理人(authorised agent)提出醫材進口許可證之申請。授權代理人依法應向CDSCO提出醫療器材進口許可證之申請(A至D所有等級)
[15]。醫療器材進口許可證之審核主要分為兩階段:
第一階段是確認境外製造廠QMS之符合性。首先,CDSCO審核授權代理人所提交的境外製造廠QMS驗證文件,包括:境外製造廠QMS之技術文件、驗證報告、最近一次之現場查核報告。如果CDSCO認定境外製造廠QMS之符合性存在疑慮時,可以要求實施評估、產品測試或檢驗程序,此時,如果CDSCO要求必須透過海外查廠的方式確認境外製造廠QMS之符合性,依法應由授權代理人負擔海外查廠之費用。
[16] 第二階段則是審核醫療器材之安全性與有效性,針對申請進口許可證之醫療器材屬於A等級或B等級之案件,授權代理人依法應向CDSCO提出原產國之自由銷售許可(free sale certificate),或是原產國實施臨床測試數據資料(或其他可供證明產品安全性與有效性之數據資料),作為CDSCO審核A等級或B等級醫療器材之安全性與有效性之證明文件。然而,若是申請進口許可證之醫療器材屬於C等級或D等級之案件,此時,CDSCO審核安全性與有效性之文件要求,會依照產品原產國是否來自先進國家而有差異。產品原產國若屬先進國家
[17],授權代理人依法提出前述自由銷售許可或原產國實施臨床測試數據資料作為證明文件即可,但若進口醫療器材之原產國為先進國家以外之其他國家,則授權代理人必須在印度實施臨床測試,以確認產品之安全性與有效性。
[18]
[1] See Drugs and Cosmetics Act - ACT No. 23 of 1940, Section 3(b)(iv).[2] Nishith Desai Associates(2017), Analysis of Medical Devices Rules, 2017, page 1.
[3] Nishith Desai Associates(2017), Analysis of Medical Devices Rules, 2017, page 2.
[4] Medical Device Rules 2017, Rule 4.
[5] Medical Device Rules 2017, Rule 20(1).
[6] 新法要求所有醫療器械產品製造地都須引入符合ISO 13485標準的品質管理系統(QMS),基於此一法規要求,印度醫療器械產業協會(Association of Indian Medical Device Industry, AIMED)與印度品質管制協會(Quality Council of India ,QCI)以及認證機構全國認證委員會(National Accreditation Board for Certification Bodies,NABCB)共同以「醫療器材品質管理體系之國際標準ISO 13485為基礎,合作制訂了印度醫療器材品質管理標準 ICMED 13485。
[7] Medical Device Rules 2017, Rule 20(4).
[8] Medical Device Rules 2017, Rule 20(5), (6).
[9] Medical Device Rules 2017, Rule 21.
[10] Medical Device Rules 2017, Rule 22(i).
[11] Medical Device Rules 2017, Rule 22(ii).
[12] Medical Device Rules 2017, Rule 21(3).
[13] Medical Device Rules 2017, Rule 23.
[14] Medical Device Rules 2017, Rule 25.
[15] Medical Device Rules 2017, Rule 34(1).
[16] Medical Device Rules 2017, Rule 34(3).
[17] 參照Medical Device Rules 2017, Rule 36(3),先進國家僅限於原產國為澳洲、加拿大、日本、歐盟國家、美國。
[18] Medical Device Rules 2017, Rule 36(4).